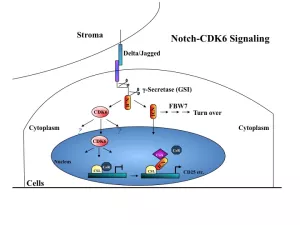
T-cell acute lymphoblastic leukemia (T-ALL) is a cancer of stem cells in the bone marrow that produce white blood cells in the body that normally fight infections. Despite recent advances in therapy, relapse is inevitable because the available therapies do not properly destroy enough cancer stem cells. Recent evidence indicates that more than half of all cases of T-ALL have constitutive active Notch1 receptor and abnormalities in PTEN and AKT pathway. However, blocking the activation of Notch1 fails to inhibit some T-ALL cells. The resistance to inhibition is due to the absence of PTEN, which results in constitutive activation of AKT. Uninhibited AKT activation leads to deregulated survival and proliferation independent of Notch signaling. Therefore, to improve therapeutic efficacy in human T-ALL, it is necessary either to inhibit both pathways or seek a common downstream mediator of the Notch1 and PI3K-AKT signaling pathways. We have found that cyclin-dependent kinase 6 (CDK6) is such a common mediator.
CDK6 regulates cell cycle progression and modulates differentiation of certain cells. It is predominantly expressed in hematopoietic cells and over-expressed in all cases of T-LBL/ALL. To clarify the role of CDK6 in cell cycle control and tumorigenesis, I have generated mice with targeted mutations in the Cdk6 gene. These knockin alleles generate hyperactive or inactive kinase subunits that may better mimic hyperactivation of CDK6 in tumor cells or model pharmaceutical intervention, respectively. Thus, these mouse models in hand are suitable to assess the relative contribution of Cdk6 kinase activity and non-catalytic activity to transformation and leukemogenesis. These animals therefore serve as useful tools for preclinical models of CDK6 function in cancer.